hybkit









- Project components:
hybkit toolkit of command-line utilities for manipulating, analyzing, and plotting hyb-format data.
The hybkit python API, an extendable documented codebase for creation of custom analyses of hyb-format data.
Integrated analysis of predicted secondary structure (fold) information for the API and command-line utilities.
Example analyses for publicly available qCLASH hybrid sequence data implemented in each of the command-line scripts and hybkit Python API.
- Hybkit Toolkit:
The hybkit toolkit includes several command-line utilities for manipulation of hyb-format data:
Utility
Description
hyb_check
Parse hyb (and fold) files and check for errors
hyb_eval
Evaluate hyb (and fold) records to identify / assign segment types and miRNAs using custom criteria
hyb_filter
Filter hyb (and fold) records to a specific custom subset
hyb_analyze
Perform an energy, type, miRNA, target, or fold analysis on hyb (and fold) files and plot results
These scripts are used on the command line with hyb (and associated "vienna" or "CT") files. For example, to filter a hyb and corresponding vienna file to contain only hybrids with a sequence identifier containing the string "kshv":
Example:
$ hyb_filter -i my_hyb_file.hyb -f my_hyb_file.vienna --filter any_seg_contains kshv
Further detail on the usage of each script is provided in the hybkit Toolkit section of this documentation.
- Hybkit API:
Hybkit provides a Python3 module with a documented API for interacting with records in hyb files and associated vienna or CT files. This capability was inspired by the BioPython Project. The primary utility is provided by a class for hyb records (HybRecord), a class for fold records (FoldRecord), and file-iterator classes (HybFile, ViennaFile, CTFile, HybFoldIter). Record attributes can be analyzed, set, and evaluated using included class methods.
For example, a workflow to print the identifiers of only sequences within a ".hyb" file that contain a miRNA can be performed as such:
#!/usr/bin/env python3 import hybkit in_file = '/path/to/my_hyb_file.hyb' # Open a hyb file as a HybFile Object: with hybkit.HybFile.open(in_file, 'r') as hyb_file: # Return each line in a hyb file as a HybRecord object for hyb_record in hyb_file: # Analyze each record to assign segment types hyb_record.eval_types() # If the record contains a long noncoding RNA type, print the record identifier. if hyb_record.has_prop('any_seg_type_contains', 'lncRNA') print(hyb_record.id)
Further documentation on the hybkit API can be found in the hybkit API section of this documentation.
- Example Analyses:
Hybkit provides several example analyses for hyb data using the utilities provided in the toolkit. These include:
Analysis
Description
Type/miRNA Analysis
Quantify sequence types and miRNA types in a hyb file
Target Analysis
Analyze targets of a set of miRNAs from a single experimental replicate
Grouped Target Analysis
Analyze and plot targets of a set of miRNAs from pooled experimental replicates
Fold Analysis
Analyze and plot predicted miRNA folding patterns in miRNA-containing hybrids
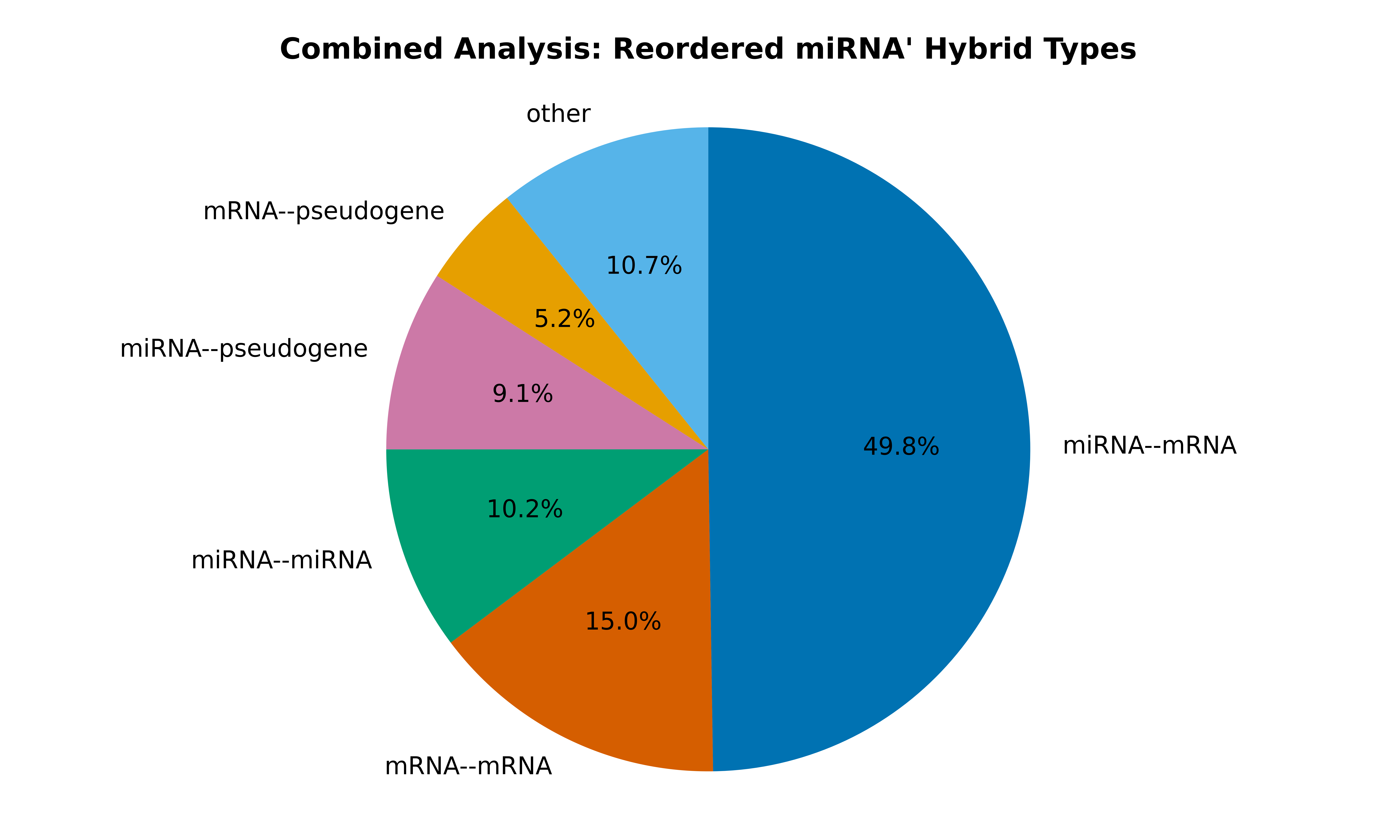
These analyses provide analysis results in both tabular and graph form. As an illustration, the example summary analysis includes the return of the contained hybrid sequence types as both a csv table and as a pie chart:
Further detail on each provided analysis can be found in the Example Analyses section of this documentation.
- Installation:
- Dependencies:
Python3.8+
matplotlib >= 3.7.1 (Hunter JD. (Computing in Science & Engineering 2007))
BioPython >= 1.79 (Cock et al. (Bioinformatics 2009))
typing_extensions <https://pypi.org/project/typing-extensions/> >= 4.8.0
- Via PyPI / Python PIP:
The recommended installation method is via hybkit's PyPI Package Index using python3 pip, which will automatically handle version control and dependency installation:
$ python3 -m pip install hybkit
- Via Conda:

For users of conda, the hybkit package and dependencies are hosted on the the Bioconda channel, and can be installed using conda:
$ conda install -c bioconda hybkit
- Via Docker/Singularity:
The hybkit package is also available as a Docker image and Singularity container, hosted via the BioContainers project on quay.io. To pull the image via docker:
$ docker pull quay.io/biocontainers/hybkit:0.3.3--pyhdfd78af_0
To pull the image via singularity:
$ singularity pull docker://quay.io/biocontainers/hybkit:0.3.3--pyhdfd78af_0
- Manually Download and Install:
Use git to clone the project's Github repository:
$ git clone git://github.com/RenneLab/hybkit
OR download the zipped package:
$ curl -OL https://github.com/RenneLab/hybkit/archive/master.zip $ unzip master.zip
Then install using python setuptools:
$ python setup.py install
Further documentation on hybkit usage can be found in this documentation.
- Setup Testing:
Hybkit provides a suite of unit tests to maintain stability of the API and script functionalities. To run the API test suite, install pytest and run the tests from the root directory of the hybkit package:
$ pip install pytest $ pytest
Command-line scripts can be tested by running the auto_test.sh script in the auto_tests directory:
$ ./auto_tests/auto_test.sh- Copyright:
- hybkit is a free, sharable, open-source project.All source code and executable scripts contained within this package are considered part of the "hybkit" project and are distributed without any warranty or implied warranty under the GNU General Public License v3.0 or any later version, described in the "LICENSE" file.
Documentation Contents: